Drug hunters, investors and partners expect three things from a modern, physics-native platform: clarity, precision and speed. PsiThera’s QUAISAR platform delivers all three. It determines whether a target is druggable and explains why, identifies what to design next and why, and accelerates discovery through closed-loop iterations that find the best molecules at scale. We leverage computational intelligence in the QUAISAR platform to turn the complexities of biology into actionable human insights, helping us create the next generation of oral medicines for diseases long dominated by injected biologics.
From Atoms to Mechanisms
PsiThera’s platform is built entirely in-house to solve one of the central challenges in drug discovery: accurately modeling biologically relevant protein motion to design oral therapeutics for validated, yet difficult, targets. Our technology integrates structural biophysics, predictive simulations, experimental data and human expertise into our unified QUAISAR (Quantum, AI and Structure-Activity Relationships) platform that can scale and adapt to any discovery program.
At PsiThera, we have built a unified computational platform from the ground up (vision, design, engineering, and optimization). Every component of our technology stack has been carefully vetted by experts in the domain-specific disciplines, including force fields, simulation engines, sampling algorithms, and AI models. We call this computational intelligence, the fusion of advanced physics, AI, and supercomputing power to understand the mechanism of biological targets in atomistic detail to design novel medicines far faster than traditional methods. This fully customized, first-principles approach gives us complete control over the scientific pipeline, enabling seamless progression from target druggability assessment to hit identification and small-molecule optimization for the most challenging protein systems. By applying quantum mechanics to compute precise molecular energetics, all-atom molecular dynamics to capture biologically relevant conformational states, and statistical thermodynamics to predict binding and selectivity, we generate a mechanistic understanding that directly informs the design of breakthrough oral therapeutics.
Machine learning models help us assess absorption, distribution, metabolism and excretion (ADME) properties and co-folding behavior, while generative AI explores vast chemical spaces to identify novel molecules. Our high-performance computing infrastructure is built specifically for these workloads. By owning the entire process end to end, we shape physics-based simulations and computational workflows around biology rather than forcing biology to fit the technology. This enables us to create bespoke in silico assays tailored to nature’s target-specific biology and our drug discovery goals.
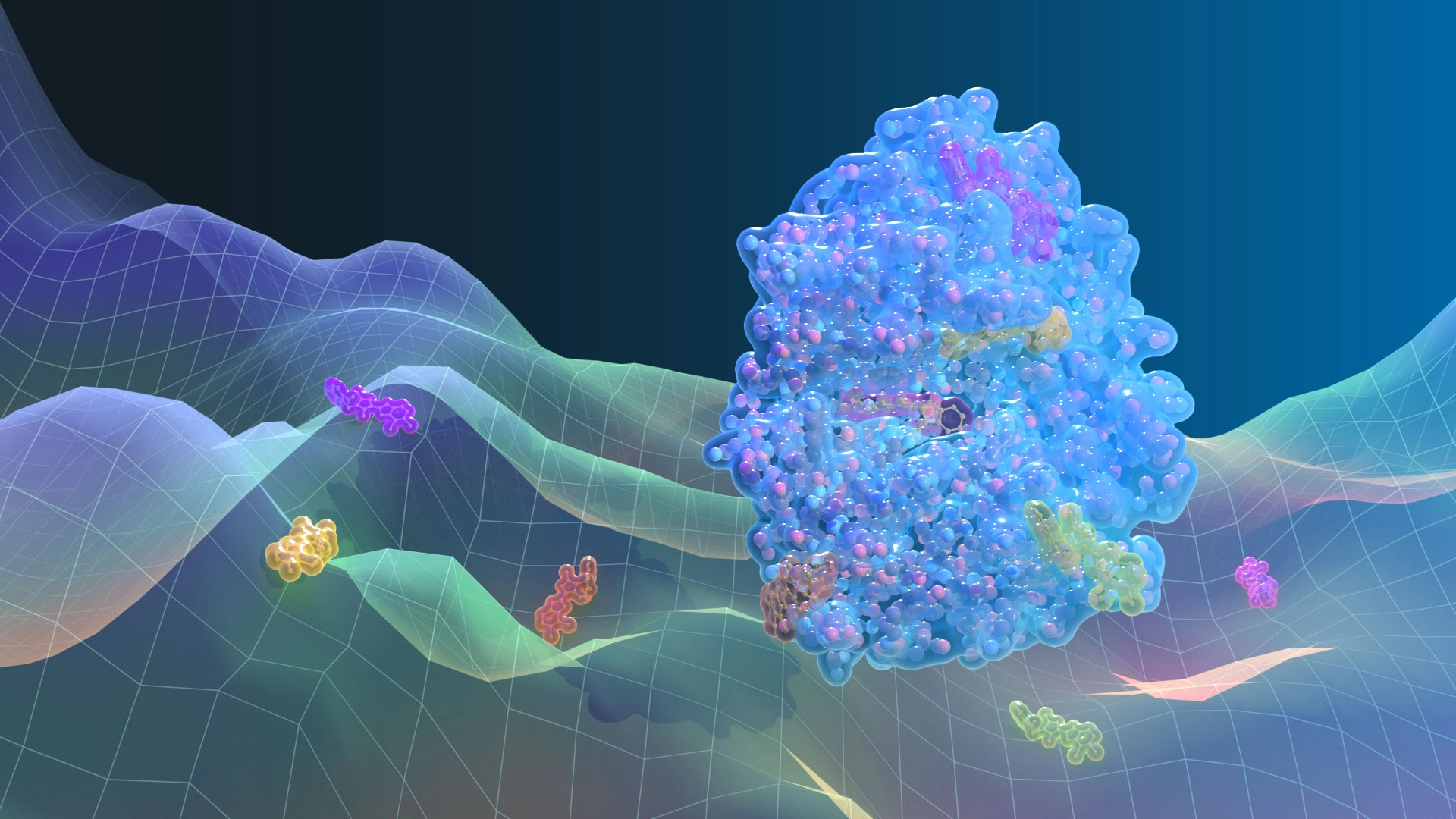
At PsiThera, we simulate biologically relevant protein motions (BRPM), enabling our team to identify cryptic pockets, transient binding sites and druggable conformations that are often undetectable in static structures. This approach enhances our understanding of activation, inhibition and signaling mechanisms. Leveraging these insights, PsiThera customizes screening strategies to efficiently discover functional and promising starting points for therapeutics targeting challenging proteins.
Computation + Wet Lab Integration
PsiThera integrates experimental structural data directly into its simulations to improve accuracy and accelerate convergence. The team combines physics with data to strengthen predictive reliability. We routinely use hydrogen-deuterium exchange mass spectrometry (HDX-MS) footprinting to set ensemble constraints, nuclear magnetic resonance (NMR) spectroscopy to capture transient dynamics and low-population states, X-ray crystallography to anchor structures, and cryogenic electron microscopy (Cryo-EM) to reveal large-scale motions and macromolecular complexes. These inputs feed into simulation protocols that refine free-energy surfaces and enhance sampling.
Every drug target has unique biology, and our platform adapts to those differences. We use in-house simulation engines and machine learning models to build custom computational assays for each project. These bespoke in silico assays address challenges such as disrupting receptor–ligand interfaces, exploring allosteric modulation and action at a distance, and analyzing membrane-associated conformational changes. We also map selectivity landscapes across protein families and evaluate synthetic tractability with chemistry-aware design. Because the QUAISAR platform is configurable at the code level, we can adjust fidelity, sampling and physics to meet project needs, delivering precision where necessary and scale where possible.
PsiVision
PsiVision is our internal data and design environment that connects scientists directly to the physics, simulations and machine-learning outputs that drive each program. It enables real-time exploration of complex simulation results, rapid visualization of free-energy landscapes and integrated maps of chemical, biological and physical properties. Scientists use human-guided prioritization with machine feedback to make transparent decisions across teams. This human-in-the-loop workflow combines computational power with expert intuition to deliver high-quality molecules quickly.
Our high-performance computing environment is built for biomolecular simulation and generative molecular design. We dedicate hundreds of GPUs to each target, run custom software tailored to project needs and use automated pipelines to handle large-scale screening. This infrastructure allows us to explore chemical and conformational space far beyond the limits of manual or empirical methods.
The PsiThera Advantage
By modeling biological motion, integrating mechanistic experimental data and building custom computational assays for each target, PsiThera achieves breakthroughs where empirical methods and AI-only platforms fall short. We harness biologically relevant protein states and computational intelligence to create oral drugs for diseases long-constrained by injectable-only approaches.